
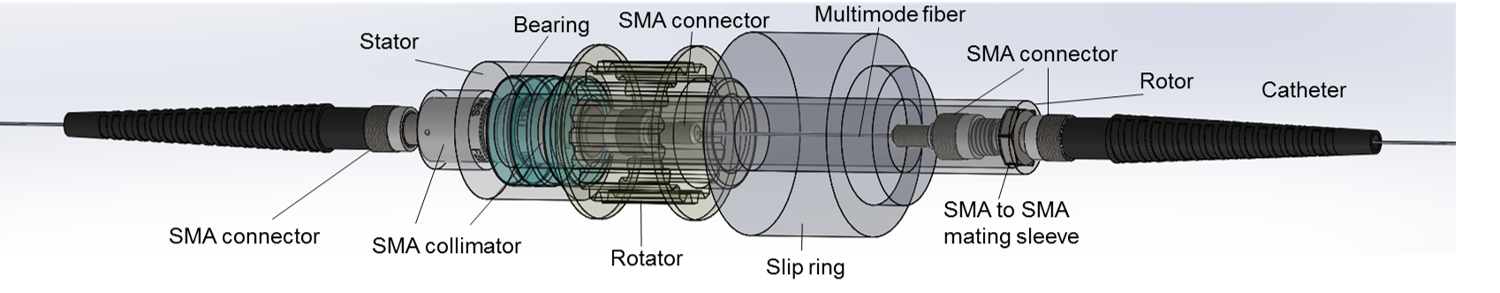
Joe Bourland Travel Award (2016)
Lu Lan of the Cheng Lab has received the Joe Bourland Travel Award to be used for his upcoming trip to SPIE Photonics West 2017.
The mission of our research program is developing label-free imaging technologies to enable improved early diagnosis and/or precise treatment of human diseases. With integrated expertise in physics, chemistry, biology, medicine and entrepreneurship, our 3D research program is devoted to
|
Development of label-free molecular spectroscopic imaging tools Discovery of new biology at single cell level Device for early molecule-based diagnosis and surgery guidance |
 |
For more information about Cheng group:
Click the link for Cheng’s Science Review Article on spectroscopic imaging https://www.sciencemag.org/content/350/6264/aaa8870.full.pdf
Click the link for Cheng’s preeminent team pitch speech https://www.youtube.com/watch?v=TtonMUcmyYY