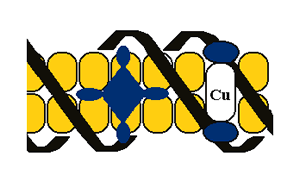
Much of our effort focuses on the chemistry of electronic excited states of transition metal complexes, especially late transition metal complexes. Because there are so many electrons in the d shell, these systems typically have vacant coordination sites and are susceptible to exciplex (excited state complex) formation. When this is the case, the excited states act as sensitive spectroscopic probes of the local environment. As an illustration of how the excited state responds to the environment, consider the platinum complex Pt(trpy)OH+, where trpy denotes 2,2':6',2"-terpyridine. This system is ordinarily non-luminescent in aqueous media, but the addition of DNA brings about important, but time-dependent, changes. The novel behavior arises because platinum terpyridine complexes bind to B-form DNA in two fundamentally different ways. In the kinetically favored adduct, the complex intercalates and becomes luminescent so long as it resides within a run of adenine-thymine (AT) base pairs. (Although guanine-cytosine (GC) base pairs shield the metal from axial interactions, guanine quenches the excited state by an electron-transfer mechanism.) Ultimately, the complex shifts into a covalently bound mode which is non-emissive so that the original emission fades out over a period of several hours. Luminescence studies are very useful for understanding the DNA-binding interactions of cationic, copper-containing porphyrins. The tetrapyridiniumyl derivative, Cu(T4), undergoes two types of non-covalent binding-- intercalation and external, or groove, binding as illustrated in the figure. The less bulky porphyrin Cu(tMe2D4) binds strictly by intercalation when excess DNA is present in solution, but it aggregates on the surface of the host if there is too little DNA present to disperse the ligand. Interestingly, the zinc(II) analogue binds in a strictly cooperative manner.
Aside from the characterization of drug interactions, luminescent metal complexes also find applications as temperature and pressure sensors. In the case of the platinum terpyridines, we find that it is possible to tinker with the orbital parentage of the excited state by introducing different types of substituents onto the terpyridine ring. Such substituent effects can change the lifetime of the excited state and the emission quantum yield by orders of magnitude. We continue to explore the effects the electronic configuration and ligand modifications have on the lifetimes, energies and reactivities of platinum terpyridines and a variety of other metal complexes.