Theoretical Chemistry Purdue
Slipchenko Group
Slipchenko Research Group

The focus of our research is on the development of theoretical and computational approaches targeting the electronic structure of extended systems, such as photosynthetic and fluorescent proteins, molecular solids, polymers, and bulk liquids.
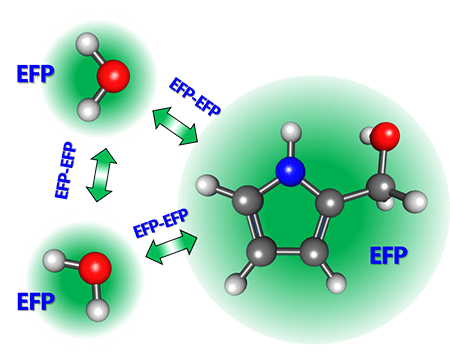
Development of EFP: Universal polarizable force field
Accurate but computationally efficient description of intermolecular interactions in extended molecular systems is one of the major challenges in computational chemistry. Our solution to this problem is the Effective Fragment Potential method (EFP), which is a universal first-principles-based parameter-free polarizable force field.
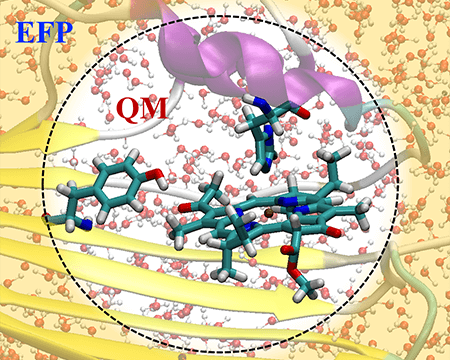
Embedding models for describing electronic structure in condensed phases
Excitation energy and electron transfer are at the heart of solar energy harvesting, both in natural photosynthetic organisms and in man-made photovoltaic devices. We learn to predict and control these phenomena by using hybrid quantum mechanics / molecular mechanics models in which classical environment is fully coupled to a quantum system.

Molecular modeling software and tools
One of the goals of our research is to develop user-friendly software for predictive modeling of properties of biological and materials systems, such as binding free energies in protein-drug complexes and lattice energies of molecular crystals. Check out our LibEFP library and iSpiEFP modeling tool!