Research in the Lipton Group
Our research efforts combine the disciplines of organic synthesis, bioorganic chemistry, and molecular modeling in research areas ranging from medicinal chemistry to peptide synthesis.

Current Research Projects
Design and Synthesis of Novel Antimicrobials
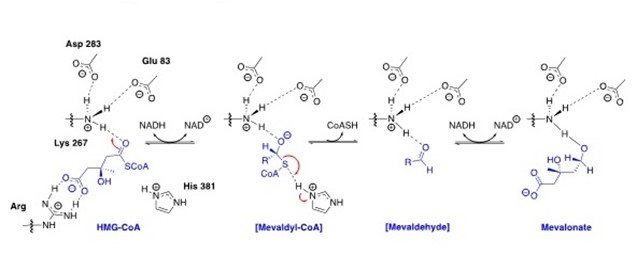 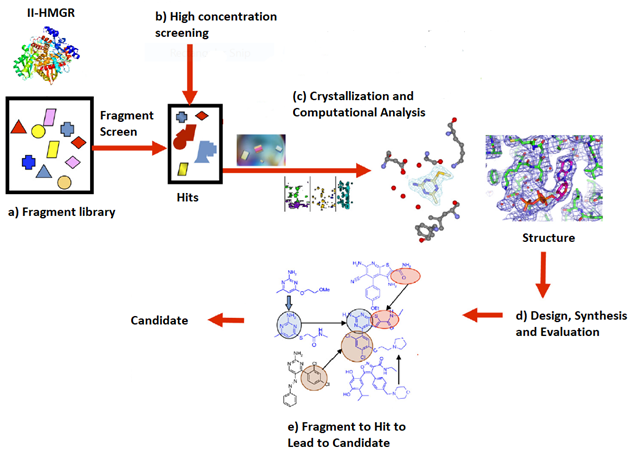 |
In 2014, the Infections Diseases Society of America (IDSA) declared multiple drug resistant bacteria to be a “substantial threat to US public health and national security”. Efforts in our lab have focused on the design, synthesis and optimization of inhibitors for gram-positive prokaryotic-specific II HMG-CoA Reductase. 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) is an enzyme that is present in both eukaryotic(I-HMGR) and prokaryotic (II-HMGR) species. It is responsible for catalyzing the reduction of HMG-CoA to mevalonate through a 4 electron oxidoreduction. In prokaryotes, mevalonate is a precursor to isopentenyl disphosphate (IPP), which is a component of undecaprenol, a lipid carrier. Undecaprenol is required for bacterial cell wall synthesis. The active site of II-HMGR consists of several different key residues compared to eukaryotic I-HMGR, making it feasible to design a molecule to selectively inhibit one in the presence of the other. To get more potent, drug-like molecule, the fragment based lead discovery approach has been adopted. With the help of the Purdue Chemical Genomics Facility, we screened 7200 compounds and selected approximately 1700 compounds based on the “Rule of 3" and a similarity screening. Selected fragments will undergo high concentration screening to identify hits. The hits will be co-crystallized and analysed computationally to design and synthesize new leads. Additionally, we are working to identify a potential secondary target of our II-HMGR inhibitors and test for cell permeability to better optimize the antimicrobial activity.
U.S. Patent 9,604,925, 2017 |
Development of a Photolabile Backbone Amide Linker
|
|
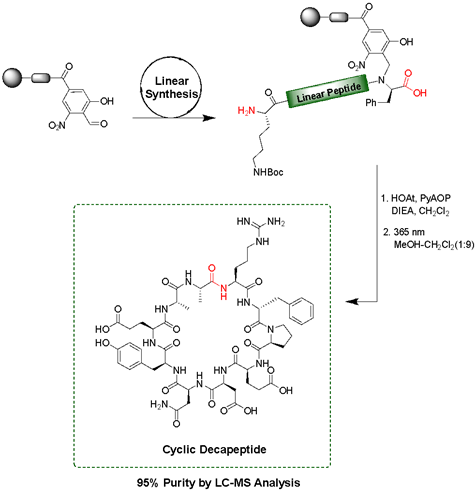
Solid-phase peptide synthesis (SPPS) is a commonly used, iterative method of building peptides that are linked at one end to an insoluble solid support. Various protecting group strategies have been used to differentiate between side chain, N-, and C-terminus functionalities. Most of these protecting groups are either acid- or base-labile, thus limiting the combinations that can be used. The peptide is attached to the solid-support through a 'linker', an organic molecule that can be cleaved in the final step under varying conditions to release the completed peptide. Backbone Amide Linkers (BALs) are commonly used as a solid-support anchor to make cyclic peptides or other C-terminal modifications. However, many of these Linkers release the peptide under the same acidic conditions used for deprotection of many side chains. We have developed a new photolabile backbone amide linker for use in the on-resin synthesis of cyclic and C-terminally modified peptides. The linker, 2-hydroxyl-4-carboxy-6-nitrobenzyl (Hcnb) is stable to strongly acidic conditions and instead releases the completed peptide through photolytic cleavage at 365 nm. We demonstrated the utility of Hcnb in the synthesis of several example cyclic peptides (5-14 residues), as well as its use in the installation of C-terminal thioesters on a variety of amino acids for the Native Chemical Ligation (NCL) technique. The peptides are typically formed in high purity (95-99%) with no HPLC purification. |

Synthesis and Structural Determination of a Macrocyclic HIV-1 Protease Inhibitor
|
|
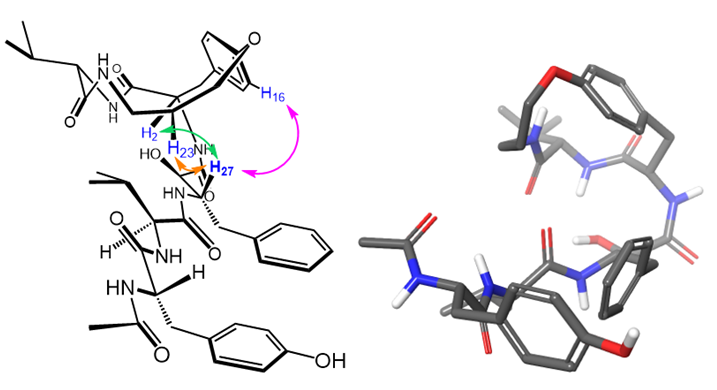
Macrocyclic, constrained templates ensure that ligands exist in a clinically desirable conformation, thereby drastically improving induced fit. Wong et al. developed macrocyclic inhibitors for the inhibition of HIV-1 protease, such as 1. After synthesizing 1, our lab determined the 3-dimensional, solution-phase structure of 1 by 2D NMR. We are now interested in using this molecular model as a ligand in docking experiments, ultimately for the design of more potent macrocyclic inhibitors of HIV-1 protease. |