Kenttämaa Labs
Analytical & Physical Organic Chemistry
Computational Research
Much of the research conducted in the Kenttämaa laboratories involves the examination of gas-phase or solution reactions by using multistage tandem mass spectrometry. These reactions often lead to the formation of unexpected products for which reaction mechanisms cannot be easily deduced. Computational chemistry allows us to explore the mechanisms of these reactions in order to improve our fundamental understanding of the chemistry of organic compounds. As an example, a potential energy surface calculated for the reaction of an ortho-benzyne analog with furan is shown below.1
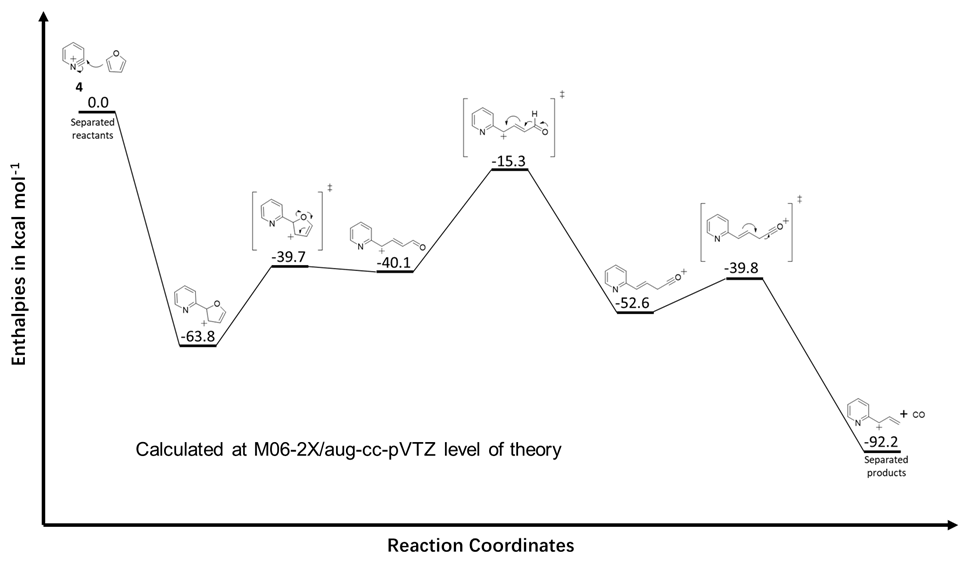
Figure 1. Calculated potential energy surface for the elimination of CO from the carbon-bound adduct formed between the 2-pyridyl cation and furan. All values are enthalpies calculated relative to the infinitely separated reactants. Note that for gas-phase ion–molecule reactions to occur, the energy levels of all transition states, reaction intermediates, and products must be below the total energy level of the system (which is the energy level of the separated reactants) because energy is not introduced into the system via solvent molecules. The stabilization of the charged reactant (here the 2-pyridyl cation) by the neutral reagent (also called solvation) provides energy to the system that can be used to overcome endothermic steps and transition states within the collision complex as long as they lie below the total energy level of the system.
These reaction mechanisms can then be used to predict how different compounds will react. For example, we have measured the rates of the bimolecular H atom abstraction reactions that take place between pyridine-based biradicals (shown below) and cyclohexane.
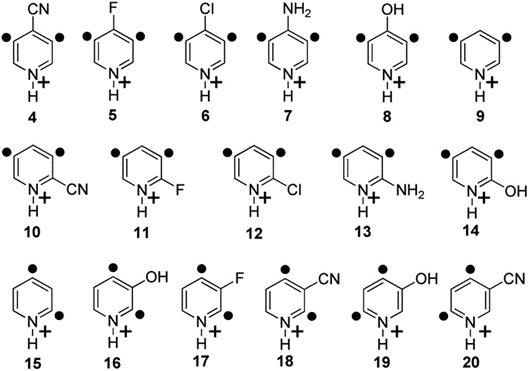
Figure 2. Structures of didehydropyridinium cations studied.
The variations in these reaction rates were rationalized based on simple thermochemical parameters, such as the biradical electron affinities, that can be calculated. In this way, we generated a model that can be used to predict whether a given biradical will efficiently abstract a H atom from cyclohexane and other hydrogen-atom donors.2
- Milton, J.R., Jankiewicz, B.J., Max, J.P., Vinueza, N.R., Kirkpatrick, L.M., Campbell, K., Gallardo, V.A., Reece, J.N. and Kenttämaa, H.I., Study on the Gas-Phase Reactivity of Charged Pyridynes. Org. Chem. 2021, 86(15), 9979-9993.
- Gao, J.; Jankiewicz, B. J.; Reece, ; Sheng, H.; Cramer, C. J.; Nash, J. J.; Kenttämaa, H. On the Factors that Control the Reactivity of meta-Benzynes. Chem. Sci. 2014, 5 (6), 2205–2215.