Research Projects
Inhibitors of the sarco/endoplasmic reticulum calcium ATPase (SERCA) as anticancer drugs
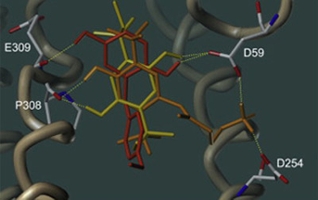 |
| Docking predicted poses of 2,5-di-tert-amylhydroquinone (yellow), 2-(4-methoxybenzyl)hydroquinone (red), and a tethered hydroquinone (orange). |
SERCA belongs to the family of P-type ATPases, which is a large class of transmembrane proteins that use the energy gained from hydrolysis of ATP to transport cations across membranes. By pumping calcium ions into intracellular storage compartments, SERCA plays a central role in calcium signaling. The ion transport activity of SERCA is inhibited by several classes of structurally unrelated compounds that include derivatives of the sesquiterpene lactone thapsigargin, the fungal metabolite cyclopiazonic acid, the antifungal drug clotrimazole, terpenolides, polyphenols, and alkylated hydroquinones. The great value of SERCA inhibitors is documented by an impressively large number of studies in the literature. Although primarily used as a powerful research tool for the study of calcium homeostasis, some SERCA inhibitors also hold promise as novel anticancer drugs. Moreover, the potential hazard to human health posed by certain SERCA inhibitors found in the environment has become a growing concern.
Our group focusses on the structural optimization of alkylated hydroquinones for effective and specific SERCA inhibition that may become potential new anticancer drugs. The small size of these compounds permits their synthesis from inexpensive starting materials in few steps, which constitutes a considerable advantage over many other inhibitors that are often natural compounds with a complex structure.
Development of ligands of the Aryl hydrocarbon Receptor (AhR) as novel agents agains breast cancer
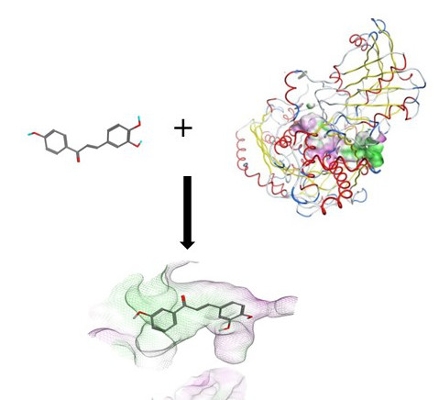
We are pursuing this project in collaboration with Adam McCluskey and other researchers at the University of Newcastle in Australia. Despite intense research efforts, breast cancer remains one of the leading causes of death for women, making the development of novel therapies for this devastating disease a top priority. Recently, the research group of Adam McCluskey has identified two new compound classes capable of killing breast cancer cells by a new mechanism. In preliminary studies, these compounds have shown great promise in that they induced breast cancer cell death at low concentrations while sparing healthy cells. In order to tap into the potential of these lead compounds and develop them into clinical candidates, optimization of their structure to improve some of their properties is required as a first step. We are contributing to this project by developing computational models capable of predicting the properties of novel compounds to be synthesized and tested by our collaborators.
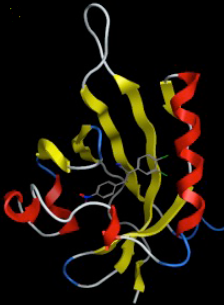 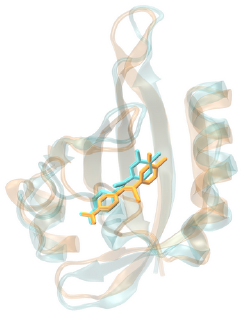 |
| Left: A ligand docked into a homology model of the AhR. Right: Ligand pose before and after 100 ns of Molecular Dynamics simulations, showing th stability of the docking-predicted pose. |
Evaluation of inhibitors of the sodium/potassium ATPase (Na/K-ATPase) for the treatment of heart failure
Even after over 200 years of use, cardiac glycosides (digitalis) are still frequently prescribed for the treatment of congestive heart failure symptoms. They exert their therapeutic effect by inhibiting the ion-transporting activity of Na/K-ATPase in the membrane of heart muscle cells. As a result of the inhibition, the transmembrane sodium concentration gradient decreases which slows the exchange of intracellular calcium ions against extracellular sodium ions. The resultant consequence is a rise in intracellular calcium concentration, which is the main cause of the so-called positive inotropic effect. The latter constitutes an increase in the contractile force of the heart muscle and cardiac output, which alleviates symptoms of congestive heart failure. Despite their effectiveness, cardiac glycosides such as digoxin or digitoxin have the disadvantage of a relatively low therapeutic index, which increases the likelihood for digitalis toxicity that can trigger life-threatening heart arrhythmias. For this reason, the availability of Na/K-ATPase inhibitors with larger therapeutic indices would be of great therapeutic value for the treatment of congestive heart failure symptoms.
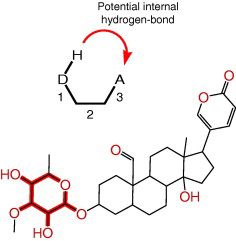 |
 |
| Illustration of then meaning of a molecular descriptor (SHBint3) that was used in a quantitative structure-activity relationship (QSAR) model capable of predicting inhibitory potencies. | Alignment of the cardiac glycoside structures used for development of a QSAR model. |
Design of Xanthine Oxidase (XO) inhibitors with dual properties against oxidatively generated stress
The enzyme XO plays an important role in purine catabolism by catalyzing the conversion of xanthine and hypoxanthine to uric acid. For decades, XO inhibition by allopurinol has been used as an effective treatment of gout, a condition caused by hyperuricemia. Disadvantages of established allopurinol-based treatments relate to the drugs' relatively low potency, requiring relatively high doses that raise the likelihood of adverse side effects. As the approval of the new XO inhibitor febuxostat in 2009 illustrates, the development of alternative XO inhibitors with novel structural scaffolds continues to be an active field of current research.
More recently, XO inhibition has been suggested to be useful for the suppression of oxidative stress in tissue caused by elevated levels of reactive oxygen species. The latter are known to cause conditions such as reperfusion injuries that can occur after surgery or ischemic events (heart attacks and strokes) when XO activity is elevated. Our goal is the development of therapeutic compounds with dual properties capable of lowering reactive oxygen species levels by inhibiting their generation by XO and of scavenging existing radicals directly.
 |
|
A chalcone-based XO inhibitor is placed in the active site of XO by molecular docking. |
Development of Cdc14 Phosphatase inhibitors as antifungal agents
This project is pursued in collaboration with Dr. Mark Hall at Purdue University. Protein phosphatase Cdc14 is essential for certain fungal species to survive and its inhibition is a promising strategy for the development of novel agents against plant pathogens. Our group’s contribution to the overall project is the computational prediction of potential inhibitors and the establishment of the molecular determinants of effective and specific fungal Cdc14 inhibition. The generated information will then be used by Mark Hall’s group for experimental testing, validation, and further development.
 |
|
Two ligands docked into the active site of Cdc14. |