Medicinal Chemistry
Current Projects
Anti-bacterial Research
The PVR group is focused on finding a cure for CDI (clostridium difficile infection) and MRSA (methicillin-resistant staphylococcus aureus) related diseases. Clostridium difficile (C. diff.) Infection (CDI) is the most common bacterial cause of hospital-acquired (nosocomial) diarrhea with an estimated 500,000 cases annually resulting in ~ 29,000 deaths in the United States alone.. Alarmingly, with the rising prevalence, severity, and death rate associated with CDI and with the infection occurring in young and healthy adults, this bacterial infection is exerting a substantial cost ($3.8 B) on the healthcare system. There is an urgent need to develop new classes of antibiotics with improved efficacy to treat CDI. Fidaxomicin is the only FDA-approved antibiotic during the past 40 years to combat CDI since Vancomycin. However, recurrence rates for fidaxomicin are still high for infections involving the hypervirulent strains (24% recurrence rate) and for patients treated for an episode of recurrent C. difficile-associated diarrhea (20% recurrence rate). One out of every five patients with CDI experienced a recurrence of the infection and 1 out of every 9 patients aged 65+ died within 30 days of diagnosis. The clostridium difficile summary data below comes courtesy of the CDC.


Hit to Lead Molecules
We have conducted a whole-cell screening assay of ~125 differently substituted small molecules prepared in our labs, against a series of clinically-relevant bacterial strains, such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecalis (VRE), vancomycin-resistant Enterococcus faecium (VRE), Clostridium difficile and Escherichia coli. Six of these molecules exhibited narrow spectrum activity against several strains of C. diff. cells. One of these (27) revealed potency comparable to the currently administered antibiotic, vancomycin, in an applicable clinical range. This new class of antibiotics has the potential for novel oral-drug development for the treatment of CDI. Our group is pursuing this hit molecule to convert it to a lead molecule.
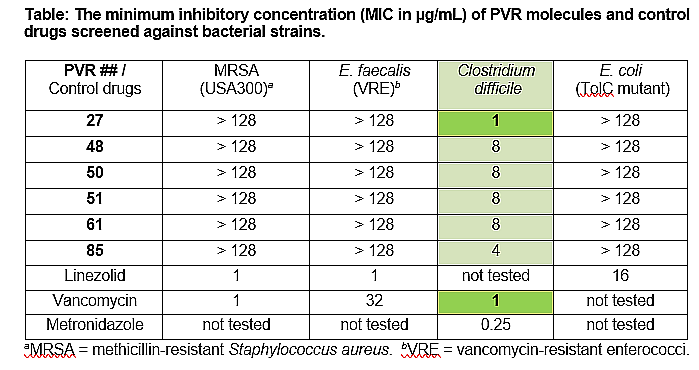
We have confirmed that these molecules can be safe drugs with completely selective antibacterial activity against C. difficile and no bio-activity against the indigenous microflora present in the GI tract! Significantly, when assayed against a human colorectal (Caco-2) cell line, these molecules revealed no toxic effect at all to mammalian cells, in vitro, even at 128 µg/mL concentration.
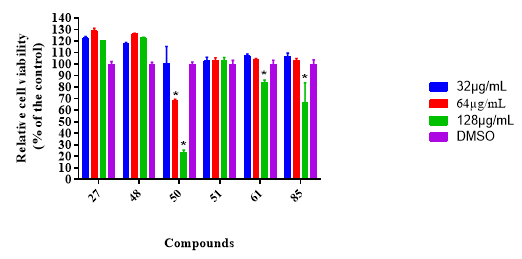
We anticipate that some of the proposed molecules may find utility as superior next generation C. difficile-specific oral drugs.
Past Projects
Anti-cancer Drug Discovery
Dictyostatin, isolated in 3.4x10-7% from marine sponges (1.3 mg from 400 Kg wet sponge) off the coast of Maldives is one of the most potent (more potent than commercial epothilones and taxanes) inducers of microtubule assembly exhibiting cytotoxicities in the nanomolar range. We reported a borane-mediated synthesis of dictyostatin in 2007 and have scaled up our reactions to prepare multimilligram quantities of this molecule. On the basis of literature reports on the mechanism of its activity and metabolism, we are completing the preparation of several analogs of dictyostatin. Following is a quotation from a 2009 Topics in Current Chemistry review entitled, Macrolide-Based Microtubule-Stabilizing Agents by Prof. Altmann of ETH, which includes our original synthesis.

The synthesis by Ramachandran et al. [189] is based on forming 8 of the 11 stereocenters of dictyostatin during the synthesis of the building blocks by pinene-mediated crotylborations [209] and another two by Myers alkylation [196] and the use of Roche ester as a starting material. To assemble the various building blocks Ramachandran et al., similar to Paterson et al. [186], first linked the C11–C17 subunit 259 by Julia olefination with the C18–C23 subunit 260 forming the northern half 261 of dictyostatin. The latter was elaborated into 262, which connected with the C1–C9 subunit 263 by substrate-controlled vinyl zincate addition [210]. The desired epimer 264 was obtained as a single isomer, which could be converted into dictyostatin (4) in five steps, including Yamaguchi macrolactonization for ring closure.
Development of NFkB-Inhibitors
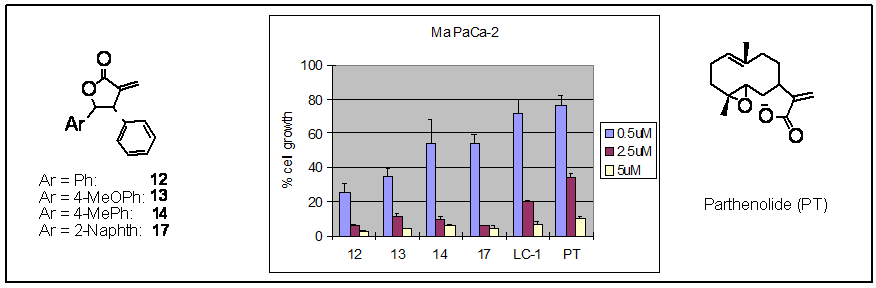
Medicinal chemists have profiled naturally occurring a-methylene-c-butyrolactones (AMGBL) for their anti-cancer properties, believed to be due to the Michael addition of intracellular thiols. However, recent studies show that parthenolide and analogs do not have a global effect on all free exofacial thiols, but rather a specific effect on 12 kDa and 22 kDa proteins. On this basis, we initiated a project on the syntheses and examination of novel AMGBLs as nuclear factor kappaB (NFKB) inhibitors.
Our initiatives in the preparation of novel functionalized allylborating agents to synthesize a-alkylidene (methylene) g-lactones of either cis or trans-stereochemistry for aryl lactones have been highly successful. Depending on the catalyst used for expediting the reaction, we have achieved both isomers in high diastereoselectivity. We are examining our novel chiral auxiliaries to achieve the asymmetric synthesis of these lactones.
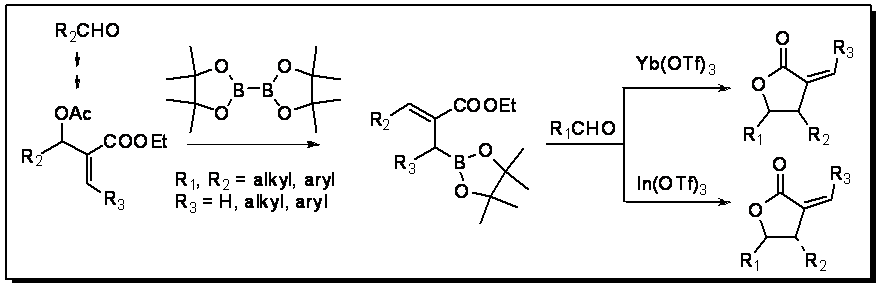
We have teamed up with Dr. C. Max Schmidt, Professor of Surgery of Indiana University School of Medicine in Indianapolis to enhance the clinical studies of the novel molecules to combat pancreatic cancer. Our analysis has identified four lactones to be as potent or slightly better inhibitors than parthenolide for the three human pancreatic cancer cells (MiaPaCa-2) examined.
Neurochemicals: CNS research
GABA (γ-aminobutyric acid) is the main inhibitory neurotransmitter in the adult mammalian central nervous system (CNS). The deficiency of GABA is related with several important neurological disorders such as Huntington’s and Parkinson’s disease, epilepsy, and other psychiatric disorders. Due to the inability of GABA to cross the blood brain barrier, their analogs have been examined for the above defeciencies. Gabapentin, Vigabatrin, Pregabalin, and Baclofen are examples of approved drugs bearing a GABA moiety. Asymmetric synthesis of GABA derivatives is an active research area in our group.
We have developed new procedures and reagents for allylboration of imines for a one-pot synthesis of g-substituted GABA derivatives.
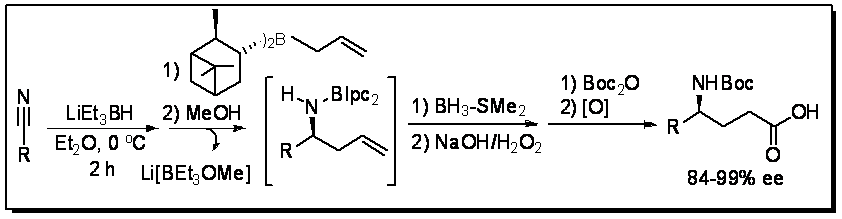
Aryl-boronic acid mediated Catalytic GABA synthesis
Our recent synthesis of GABA derivatives via a Rh catalyzed addition of boronic acids to sulfinylimines.
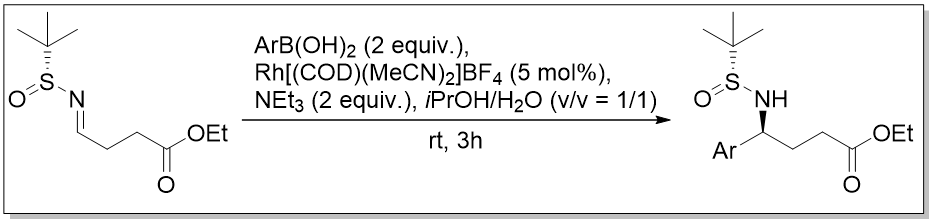
N-metallo fluoro-imines, prepared from nitriles, were transformed to g-aminobutyric acids, an important component of our on-going collaborative research in nervous system disorders. In our studies involving innovative approaches to target excitatory or inhibitory amino acids, we ascertained a previously un-described role for some of our F-GABA derivatives. We demonstrated that 10 mM of 3,4-DIF robustly potentiated GABA stimulation at the GABA-A receptors in HEK cells expressing the a1b2g2 subunits, without appreciable agonist activity, accessing the benzodiazepine binding site at the a1/g1 subunit interface of the GABA-A receptor in a dose-dependent fashion. The EC50 for potentiation of 1 mM GABA agonist activity at the a1b2g2 receptor by 3,4-DIF is 256 nM, and the maximum potentiation of current is ~42%. Notably, 3,4-DIF did not have any appreciable effect on GABA-stimulated currents in those GABA-A receptors lacking the g subunit, also characteristic of the commercial drug zolpidem (Ambien). Further clinical evaluations are under way.