The Ghosh Laboratories:
The Home of the Backbone Binding, Bioactive Natural Product Synthesis, and Nature-Inspired Molecular Design for Today's Medicine
Development of Asymmetric Synthetic Methods
We have developed a variety of new and useful asymmetric reactions based upon various intermolecular or intramolecular metal chelation. These include syn and anti specific aldol reactions, multicomponent reactions, inter- and intramolecular Diels-Alder reactions, conjugate additions, asymmetric protonation and reductions.
Development of Multicomponent reactions
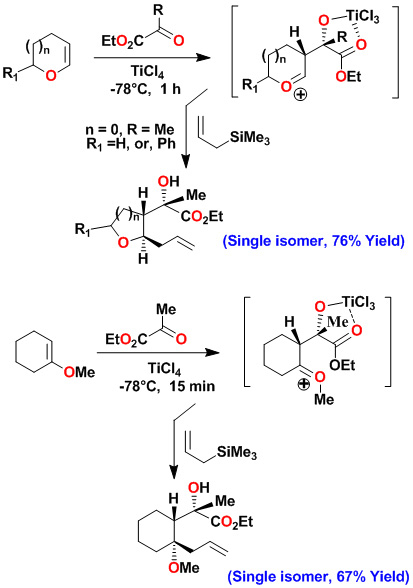
A variety of substituted pyrrolidines have been prepared in a one-pot operation. An asymmetric multicomponent reaction of an optically active dihydrofuran with ethyl pyruvate provided our presumed oxocarbenium ion which was reacted with purine and pyrimidine bases to provide modified nucleosides in high diastereoselectivity and good yields.
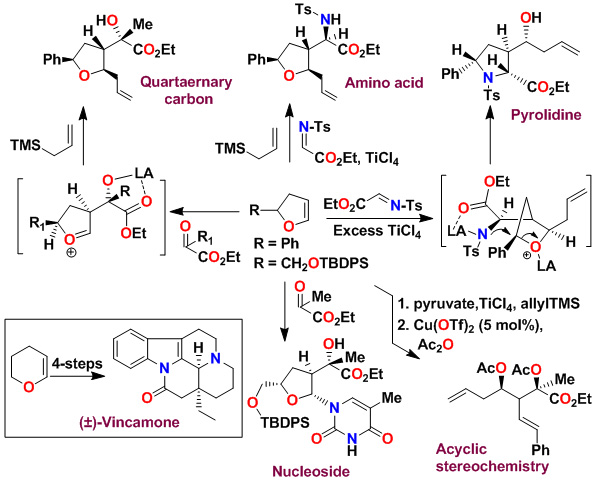
Key references: Ghosh, A. K.; Kass, J. ChemComm. 2010, 46, 1218-1220; Ghosh, A. K., et. al. Tetrahedron Asymm. 2008, 19, 1020-1026; Ghosh, A. K.; Xu, C.; Kulkarni, S. Fanwick, P. E. Organic Letters 2006, 8, 4509-11. Ghosh, A. K.; Xu, C.; Kulkarni, S.Organic Letters 2005, 7, 7-10; Ghosh, A. K.; Kawahama, R. J. Org. Chem. 2000, 65, 5433; Ghosh, A. K.; Kawahama, R. Tetrahedron Letters, 1999, 40, 1083; Ghosh, A. K.; Kawahama, R.; Wink, D. Tetrahedron Letters 2000, 44, 8425; Ghosh, A. K.; Kawahama, R. Tetrahedron Letters, 1999, 40, 1083.
Asymmetric Anti-Aldol Reactions
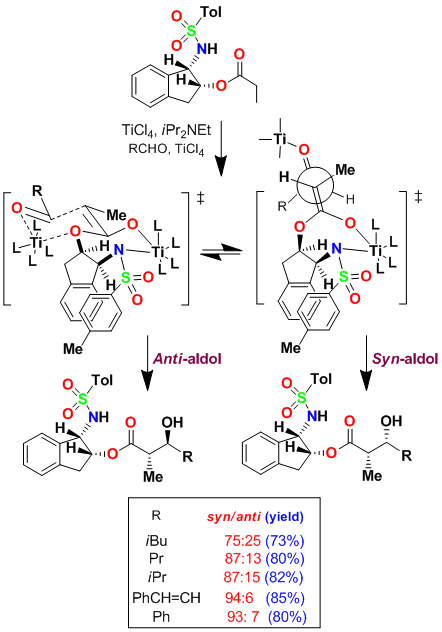
We have developed highly diastereoselective ester-derived titanium enolate-based anti-aldol and syn-aldol reactions. The titanium enolate was generated with TiCl4 and i-Pr2NEt at 0°C to 23°C. These conditions provided the Z-enolate, which upon reacting with various monodentate aldehydes complexed with TiCl4 afforded anti-aldol products with excellent diastereoselectivity and isolated yields. These reactions were extensively used in the synthesis of variety of bioactive molecules.
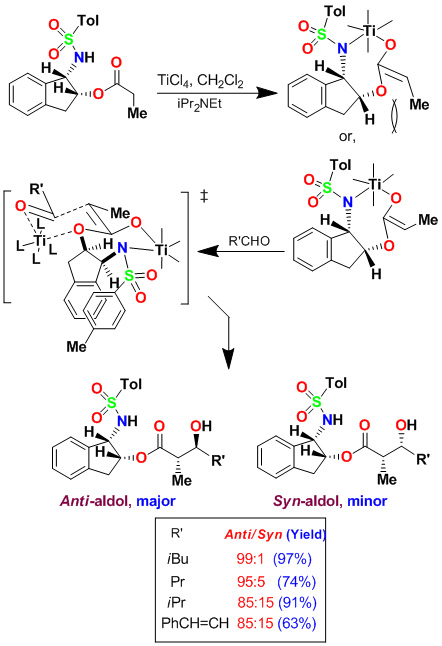
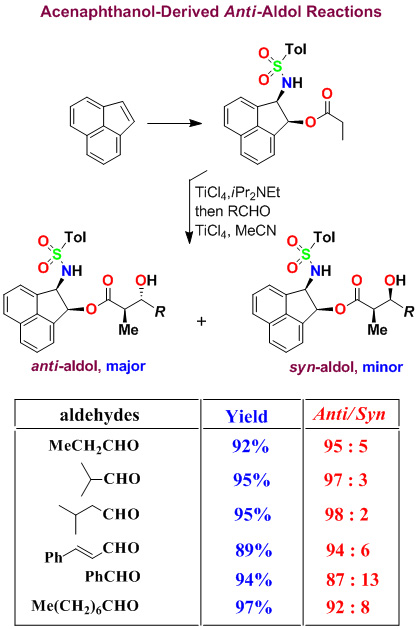
Asymmetric Syn-Aldol Reactions
Based upon the proposed transition-state assembly for the anti-aldol reaction, we postulated that a bidentate alkoxyaldehyde would adopt a transition state so that the ether oxygen could effectively donate its lone pair to the vacant d-orbital of titanium and the alkoxy aldehyde side chain would be oriented pseudo-axially, giving rise to the syn-aldol product selectively. Indeed, reaction of benzyloxyacetaldehyde and benzyloxypropionaldehyde proceeded with excellent diastereoselectivity. The aldol reaction with benzyloxybutyraldehyde provided slightly reduced syn-diastereoselectivity (dr 94:6). Aldol reactions of chloroacetate with the bidentate aldehydes provided aldol products with high syn-diastereoselectivity and yields.
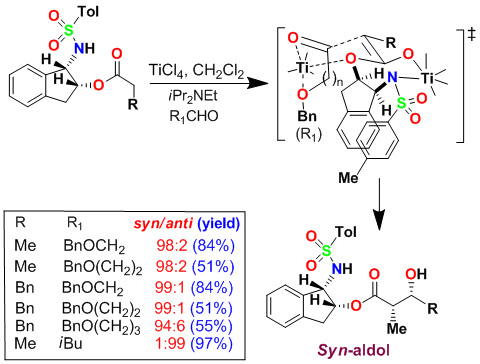
Based upon our proposed highly chelated transition state, we further speculated that a a-chiral center on the indane ring may not be required for syn-diastereoselectivity with bidentate aldehydes. We therefore investigated amino acid-derived chiral sulfonamides. As shown, a phenylalaninol-derived auxiliary provided the desired aldol products with excellent syn-diastereoselectivity (dr 98:2) with alkoxyaldehyde.
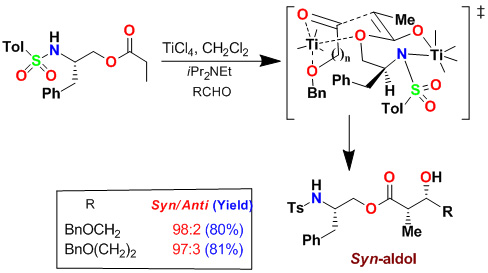
As shown, activation of monodentate aldehydes with TiCl4 and other Lewis acids were necessary to observe anti-diastereoselectivity. We have subsequently investigated the effect of increasing quantities of TiCl4 on the syn:anti product ratio and reaction yields. Interestingly, when cinnamaldehyde was complexed with 3 equiv of TiCl4, there was a dramatic reversal of diastereoselectivity. Reaction of 2 equiv of cinnamaldehyde, precomplexed with 5 equiv of TiCl4, provided excellent yield (95%) and excellent syn-diastereoselectivity (dr: 95:5). As shown, the generality of this reaction was examined with a variety of aldehydes.
Key references: Ghosh, A. K.; Dawson, Z. L. Synthesis 2009, 2992-3003; Ghosh, A. K.; Shevlin, M. Modern Aldol Reactions, WILEY-VCH, 2004, 63-121; Ghosh, A. K.; Kim, J.-H. Organic Letters 2003, 6, 1063; Ghosh, A. K.; Bischoff, A. Organic Letters 2000, 2, 1573; Ghosh, A. K.; Fidanze, S.; Senanayake, C. H. Synthesis, 1998, 937; Ghosh, A. K.; Fidanze, S.; Onishi, M. Tetrahedron Lett. 1997, 38, 7171; Ghosh, A. K.; Onishi, M. J. Am. Chem. Soc., 1996, 118, 2527.
Asymmetric Catalytic Diels-Alder reactions
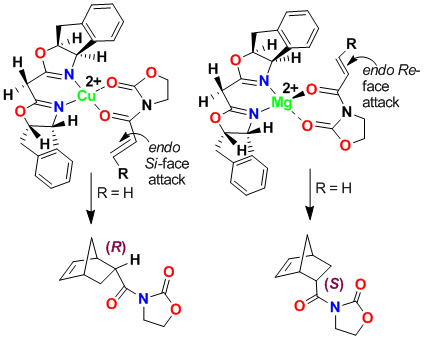
We have investigated cis-aminoindanol-derived bis(oxazoline)-metal-catalyzed asymmetric reactions. The C2-symmetric chiral bis(oxazoline) ligands have now been developed as privileged ligands for a variety of asymmetric transformations. In an effort to improve Phe-Box-Cu(II)-catalyzed Diels-Alder reactions, we speculated that the cis-aminoindanol-derived bis(oxazoline) may well serve as a conformationally constrained alternative to the Phe-Box ligand. In this context, we first prepared aminoindanol-derived bis(oxazoline) ligands by condensation of the imidate salt with optically active aminoindanol. We have prepared a variety of metal-bis(oxazoline) complexes as catalysts for asymmetric Diels-Alder and hetero Diels-Alder reactions. As shown, Cu(II)-catalyzed reactions with acryloyl-N-oxazolidinone proceeded with excellent endo/exo selectivity as well as endo-enantioselectivity (up to 99% ee), in contrast to Phe-Box-Cu(II)-catalyzed reactions (30% ee).
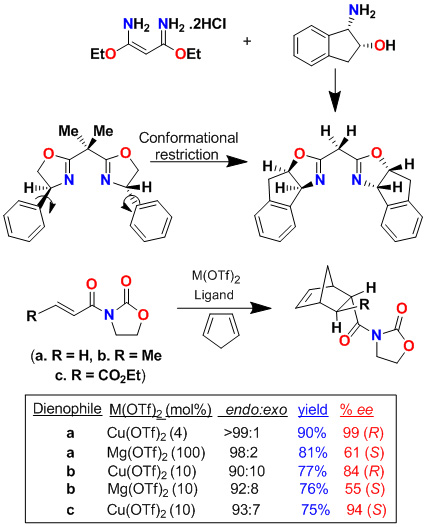
Furthermore, we have investigated cationic aqua complexes derived from INDA-Box and Cu(ClO4)2·6H2O or Ni(ClO4)2·6H2O, which provided Diels-Alder products in excellent yield and enantioselectivity (up to 98% ee). Interestingly, we have observed a reversal of enantioselectivity with Mg(II)-catalyzed reactions (55-65% ee). These results were rationalized using a square planar model of the Cu(II)-ligand complex and a S-cis conformation of the dienophile as shown.
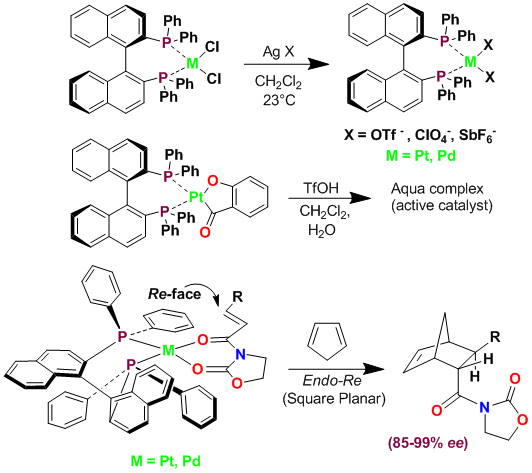
We have demonstrated that some cationic Pt(II) and Pd(II) chiral bisphosphine complexes are highly effective catalysts for the enantioselective Diels-Alder reaction of cyclopentadiene with various bidentate dienophiles. With appropriate counterions, the reaction proceeds with excellent endo/exo selectivity, endo enantioselectivity (up to 99% ee) and isolated yields.
Asymmetric Hetero Diels-Alder Reactions
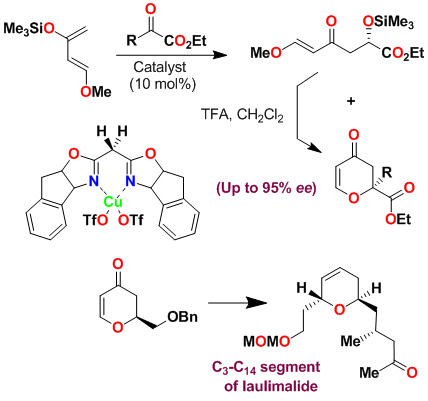
We have investigated asymmetric hetero-Diels-Alder reactions using bis(oxazoline)-metal complexes with Danishefsky's diene and bidentate aldehydes such as glyoxylate ester, 1,3-dithiane-2-carboxaldehyde and benzyloxyacetaldehyde. The reaction proceeded in a stepwise manner via a Mukaiyama aldol reaction followed by ring closing with trifluoroacetic acid (TFA) to provide dihydropyranone. Reactions of benzyloxyacetaldehyde provided dihydropyranone in 76% yield and 95% ee. This was converted to the C3-C14 segment of laulimalide. Our investigation on a-keto esters with Danishefsky's diene provided dihydropyranone derivatives in good to excellent yield and high enantioselectivity (up to 99% ee). This methodology was used to construct a quaternary carbon center for the synthesis of marine natural product (-)-malyngolide.
Key references: Ghosh, A. K.; Shirai, M. Tetrahedron Letters 2001, 42, 6231; Ghosh, A. K.; Matsuda, H. Organic Letters 1999, 1 2157; Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Tetrahedron: Asymmetry, 1998, 9, 1-48; Ghosh, A. K.; Cho, H.; Cappiello, J. Tetrahedron : Asymmetry, 1998, 9, 3688; Ghosh, A. K.; Mathivanan, P.; Cappollo, J. Tetrahedron: Asymmetry, 1996, 7, 2165.
Asymmetric Reductive Aldol Reactions
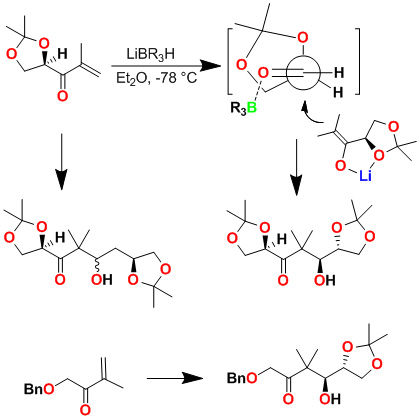
We have developed a highly diastereoselective asymmetric reductive aldol reaction. As shown below, an aldol reaction of the enolate generated by addition of l-selectride to an enone and isopropylidene-D-glyceraldehyde in ether at -78°C provided only one diastereomer, in 70% yield. Reaction with chiral isopropylidenebutyraldehyde however, provided a 1:1 mixture of diastereomers. The corresponding reaction with isovaleraldehyde also provided a 1:1 mixture of diastereomers, suggesting that the a-chirality of the isopropylidene-d-glyceraldehyde is responsible for the diastereoselectivity as proposed in the stereochemical model. Indeed, reaction with an achiral enone and isopropylidene-D-glyceraldehyde provided a single isomer. This asymmetric reductive aldol reaction was utilized in the synthesis of peloruside A and B.
Key publications: Ghosh, A. K. J. Org. Chem. 2010, 75, 2-10; Ghosh, A. K.; Kass, J.; Anderson, D. D.; Xu, X.; Marian, C. Organic Letters 2008, 10, 4811-4814; Ghosh, A. K.; Xu, X.; Kim, J.-H.; Xu, C.-X. Organic Letters 2008, 10, 1001-1004.