Anionic Control of Nitrene Electronic Structure

One of the focuses of our work is on the investigation of the electronic structures of aromatic nitrenes, and, in particular, how to modify the relative energies of the electronic states. Nitrenes are a fascinating class of electron-deficient and highly reactive intermediates, with electronic structures nominally similar to their carbon analogs, carbenes. Of particular interest is phenylnitrene, which has been the subject of extensive experimental and computational studies. However, while carbenes and nitrenes share some similarities, they exhibit very different reactivity.1-3 For example, phenylcarbene in solution readily forms adducts with alkenes and inserts into C-H bonds, whereas phenylnitrene (PN) gives mostly polymeric tar.4 The difference in reactivity can be attributed to differences in electronic structures, in particular the structures of the lowest-energy singlet state. Both PN and phenylcarbene areknown to be ground-state triplets,1,5-7 but whereas the lowest energy singlet state in phenylcarbene is a closed-shell, σ2 state that is about 2 – 5 kcal/mol6,8 above the triplet, the singlet in PN is an open-shell, σπ state, with a singlet-triplet splitting of 14.9 kcal/mol7,9-11 and the σ2 state predicted to be approximately 30 kcal/mol higher than the triplet.7,10
In an effort to tame the reactivity of aromatic nitrenes, we suggested the use of quinonimides as nitrene-analogs with low-lying closed-shell singlet electronic states.12 The electronic structure and energy of the lowest energy singlet state is important because the singlet nitrene generated by nitrogen loss from the corresponding azide subsequently undergoes intersystem crossing (ISC) to the triplet. Rates of ISC by spin-orbit coupling depend not only on the energy of the electronic states, but also on their structures, because effective ISC requires an orbital transition to create the torque needed to change the angular momentum of the electron.13 Consequently, favorable ISC requires a change in orbital occupancy in addition to change in the electron spin. For PN, ISC from the σπ singlet state to the triplet is slow because it does not involve a change in orbital angular momentum to accompany the change in electron spin.13 Instead of ISC, the open-shell singlet state of PN can also undergo unimolecular rearrangement, as shown in Scheme 1.5 In solution, the singlet can ring-expand to form the cyclic ketenimine, K, which can react further. In the gas phase, the same singlet undergoes ring contraction to a cyano-substituted 5-membered ring.14-19 The products formed in the reaction depend on the relative rates of the competing processes. For singlet PN in solution at temperatures >180K, ring-expansion occurs faster than ISC, which accounts for the formation of polymeric products. Below 180K, ISC is favored to form the triplet state, which can then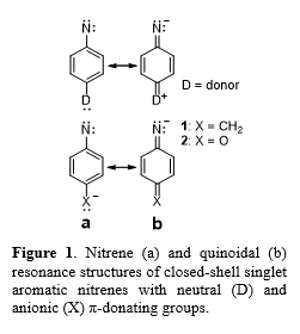 undergo bimolecular reactivity. In contrast, singlet phenylcarbene does not undergo unimolecular rearrangement because ISC is faster under all conditions. One strategy for harnessing the reactivity of phenylnitrene is to increase the rate of ISC to prevent unimolecular rearrangement. For example, introducing strong π-donating groups, such as –OCH3 and –N(CH3)2, in the para position of phenylnitrene has been found to increase the rate of ISC by factors of about 150 and 2500, respectively.16 Nominally, the increase in the rate of ISC can be attributed to stabilization of the closed-shell singlet state of the nitrene by resonance, as shown in Figure 1, as ISC should be more favorable for the closed-shell singlet state. Computational studies predict that, indeed, addition of a π-donating group at the para position of phenylnitrene stabilizes the closed-shell singlet relative to the triplet, and can increase the rate of ISC.20 However, the extent of stabilization is not sufficient to overcome the nearly 30 kcal/mol energy gap between the states in the unsubstituted system, nor even the difference between the closed-shell and open-shell singlet states, which is approximately 15 kcal/mol.20 The reason strong π-donors fail to lower the energy of the closed-shell singlet enough to change the state ordering in aromatic nitrenes is due in part to the diminishing returns that the singlet encounters because of increased zwitterionic character, as shown in the quinoidal structure (structure b) in Figure 2.16
undergo bimolecular reactivity. In contrast, singlet phenylcarbene does not undergo unimolecular rearrangement because ISC is faster under all conditions. One strategy for harnessing the reactivity of phenylnitrene is to increase the rate of ISC to prevent unimolecular rearrangement. For example, introducing strong π-donating groups, such as –OCH3 and –N(CH3)2, in the para position of phenylnitrene has been found to increase the rate of ISC by factors of about 150 and 2500, respectively.16 Nominally, the increase in the rate of ISC can be attributed to stabilization of the closed-shell singlet state of the nitrene by resonance, as shown in Figure 1, as ISC should be more favorable for the closed-shell singlet state. Computational studies predict that, indeed, addition of a π-donating group at the para position of phenylnitrene stabilizes the closed-shell singlet relative to the triplet, and can increase the rate of ISC.20 However, the extent of stabilization is not sufficient to overcome the nearly 30 kcal/mol energy gap between the states in the unsubstituted system, nor even the difference between the closed-shell and open-shell singlet states, which is approximately 15 kcal/mol.20 The reason strong π-donors fail to lower the energy of the closed-shell singlet enough to change the state ordering in aromatic nitrenes is due in part to the diminishing returns that the singlet encounters because of increased zwitterionic character, as shown in the quinoidal structure (structure b) in Figure 2.16
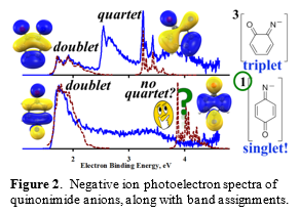
Additional insight into the electronic structure of 2 was reported recently, obtained from negative ion photoelectron spectroscopy studies, carried out in collaboration with Prof. X. Wang at Pacific Northwest Laboratories. The photoelectron spectra of 2 and its ortho-isomer (2o) are shown in Figure 2. Perhaps the most significant observation regarding the spectrum of 2 is its simplicity. There is only one intense feature, which is readily assigned to formation of the doublet ground state of the iminyl radical. The lack of a corresponding quartet state, which is predicted to be present in high relative intensity, is significant in that it indicates that the ion has a singlet ground state. In order to obtain the correct prediction with electronic structure theory requires using a very high level of theory that accounts for the dynamic correlation within multi-reference wave function of the singlet state and a basis set augmented with diffuse functions to account for the presence of negative charge.
In contrast, the spectrum for 2o contains features for both doublet and quartet formation, indicating a triplet state for the ion. The spin-state assignment is consistent with electronic structure calculations, which predict a singlet-triplet splitting of about 9 kcal/mol, and gas-phase reactivity studies, which indicate open-shell character.
Nitrenes: Applications in chemical reactivity

The electronic structure of 1o as a triplet nitrene with a closed-shell singlet ground state resembles that of a carbene. As such, it may be possible that the nitrene undergoes reactivity that resembles that of a carbene. Figure 3 shows the electrospray ionization (ESI) mass spectrum of the photochemical reaction of o-azidophenoxide in the presence of but-3-en-2-ol. The most significant product observed in the spectrum is at m/z 178, which corresponds to addition and loss of N2. This is the mass that corresponds to that of the aziridine that would be formed upon trapping the nitrene! The NMR spectrum of the product is also consistent with that expected for the aziridine, but is not unequivocal. We are currently working to obtain a crystal structure to confirm the assignment. Consequently, it appears promising that we have been able to carry out chemical trapping of the nitrene to form an aziridine. Although aziridines can be formed from organometallic nitrene-analogs, the direct trapping of nitrenes to form aziridines generally does not occur. As stated by Matt Platz, “…phenylnitrene as a reagent is never described with complimentary adjectives.”1 Ion 1o may be the exception!
Similar attempts to trap 1p to form an aziridine product have been unsuccessful. Haas and co-workers22 have previously shown that photolysis of p-azidophenoxide in aprotic solvents leads to the formation of a diazene product resulting from dimerization of the nitrene, which is evidence of nitrene formation. However, the nitrene has not been trapped to form an aziridine.
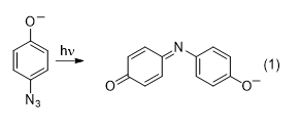
An unusual result is obtained when the photolysis is carried out in protic solvent. Instead of getting a nitrene-based product, the product is indophenol (eq 1).22 The indophenol product in the reaction is surprising, and the mechanism is poorly understood. Although Haas and co-workers22 proposed a mechanism based on the observed products, additional studies carried out in our lab have provided more insight. While the work is still on-going, our mechanistic studies are facilitated by the fact that we are able to carry out real-time monitoring of the reaction by using ESI mass spectrometry, where we can observe the loss of reactant and formation of product, as well as other ionic intermediates that are present. Our results suggest that the reaction likely results from single-electron transfer (SET). More detailed mechanistic studies, being carried out by undergraduate students, are still underway.
References
-
Platz, M. S. Comparison of Phenylcarbene and Phenylnitrene. Acc. Chem. Res. 1995, 28, 487-492.
-
Horner, L.; Christmann, A. Nitrenes. Angew. Chem. Int. Ed. 1963, 2, 599-608.
-
Belloli, R. Nitrenes. J. Chem. Edu. 1971, 48, 422-426.
-
Meijer, E. W.; Nijhuis, S.; van Vroonhoven, F. C. B. M. Poly-1,2-azepines by the Photopolymerization of Phenyl Azides. Precursors for Conducting Polymer Films. J. Am. Chem. Soc. 1988, 110, 7209-7210.
-
Borden, W. T.; Gritsan, N. P.; Hadad, C. M.; Karney, W. L.; Kemnitz, C. R.; Platz, M. S. The Interplay of Theory and Experiment in the Study of Phenylnitrene. Acc. Chem. Res. 2000, 33, 765-771.
-
Gronert, S.; Keeffe, J. R.; More O'Ferrall, R. A. Correlations between Carbene and Carbenium Stability: Ab Initio Calculations on Substituted Phenylcarbenes, Nonbenzenoid Arylcarbenes, Heteratom-Subtituted Carbenes, and the Corresponding Carbocations and Hydrogenation Products. J. Org. Chem. 2009, 74, 5250-5259.
-
Winkler, M. Singlet-Triplet Energy Splitting and Excited States of Phenylnitrene. J. Phys. Chem. A 2008, 112, 8649-8653.
-
Geise, C. M.; Hadad, C. M. Computational Study of the Electronic Structure of Substituted Phenylcarbene in the Gas Phase J. Org. Chem. 2000, 65, 8348-8356.
-
Travers, M. J.; Cowles, D. C.; Clifford, E. P.; Ellison, G. B. Photoelectron Spectroscopy of the Phenylnitrene Anion. J. Am. Chem. Soc. 1992, 114, 8699-8701.
-
Wenthold, P. G. Spin-State Dependent Radical Stabilization in Nitrenes: The Unusually Small Singlet-Triplet Splitting in 2-Furanylnitrene. J. Org. Chem. ASAP, 77, 208-214.
-
Wijeratne, N. R.; Da Fonte, M.; Ronemus, A.; Wyss, P. J.; Tahmassebi, D.; Wenthold, P. G. Photoelectron Spectroscopy of Chloro-Substituted Phenylnitrene Anions. J. Phys. Chem. A 2009, 113, 9467-9473.
-
Rau, N. J.; Welles, E. A.; Wenthold, P. G. Anionic Substituent Control of the Electronic Structure of Aromatic Nitrenes. J. Am. Chem. Soc. 2013, 135, 683-690.
-
Salem, L.; Rowland, C. The Electronic Properties of Diradicals. Angew. Chem. Int. Ed. 1972, 11, 92-111.
-
Alekseyev, P. V.; Romanova, I. V.; Shakirov, M. M.; Godovikova, T. S. p-Azidophenyl phosphate is a photoactivatable phosphorylating reagent and p-benzoquinone monoimine precursor. J. Photochem. Photobiol., B 2001, 65, 39-46.
-
Modestov, A. D.; Gun, J.; Savotine, I.; Lev, O. On-line electrochemical-mass spectrometry study of the mechanism of oxidation of N,N-dimethyl-p-phenylenediamine in aqueous electrolytes. J. Electroanal. Chem. 2004, 565, 7-19.
-
Gritsan, N. P.; Platz, M. S. Kinetics and Spectroscopy of Substituted Phenylnitrenes. Adv. Phys. Org. Chem. 2001, 36, 255-304.
-
Wang, Z.; Li, X.; Wu, Y.; Tang, Y.; Ma, S. Spectroelectrochemistry for a coupled chemical reaction in the channel cell Part II. Kinetics of hydrolysis and the absorption spectrum of p-benzoquinoneimine. J. Electroanal. Chem. 1999, 464, 181-186.
-
Song, Y. Theoretical studies on electrochemistry of p-aminophenol. Spectrochim. Acta, Part A 2007, 67A, 611-618.
-
Song, Y. Z.; Xie, J. M.; Song, Y.; Ye, Y. Theoretical and experimental studies of the electrochemistry of p-aminophenol on a golden electrode. Russ. J. Phys. Chem. A 2007, 81, 1669-1676.
-
Liu, X.; Tao, Z.; Li, G. Determination of reaction rate constant of platinum-catalyzed oxidation of p-aminophenol. Shandong Jiancai Xueyuan Xuebao 1997, 11, 207-213.
-
Rau, N. J.; Welles, E. A.; Wenthold, P. G. Anionic Substituent Control of the Electronic Structure of Aromatic Nitrenes. J. Am. Chem. Soc. 2013, 135, 683-690.
-
Geiger, U.; Haas, Y.; Grinstein, D. The photochemistry of an aryl pentazole in liquid solutions: The anionic 4-oxidophenylpentazole (OPP). J. Photochem. Photobiol. A: Chem. 2014, 277, 53-61.