Theoretical Chemistry Purdue
Slipchenko Group
Theory and development
Calculations in the condensed phase still remain a major challenge to the theoretical community. The increased number of nuclear and electronic degrees of freedom makes accurate ab initio calculations on a condensed phase system unfeasible long before the system can approach the bulk. One general approach to this type of problem is to separate a system into two parts, such that one (active) part is treated by quantum mechanical (QM) techniques, and the other, usually larger, part is calculated by using classical (molecular) mechanics (MM).
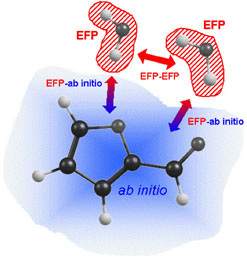
Traditionally, the MM part in QM/MM is included through parameterized force fields. A drawback of such an approach is the dependence on fitted parameters for a chosen force field, such that different parameterizations may be optimal for different problems and the best parameters are often not well defined. In order to overcome this problem, we employ the Effective Fragment Potential (EFP) method for the MM part. In the EFP, each solvent molecule is represented by a model potential with a set of parameters determined from a preparatory ab initio calculation. The uniqueness of the EFP method is that all parameters are derived from first principles, i.e., the method is free of parameter fitting. Through its force field, the EFP fragments can interact with each other and with ab initio components. Using the EFP method results in an accurate and first-principles-based description of the MM part and couplings between the QM and MM subsystems. Our EFP and QM/EFP methods are available in the GAMESS and Q-Chem electronic structure packages.
Development of the EFP method
Book chapters about EFP: 2007 (PDF), 2016 (PDF)
Review of recent developments (PDF)
Short-range damping functions in EFP: exponential-based (PDF) and overlap-based (PDF)
Coarse-graining of EFP (PDF)
EFP implementation in Q-Chem: 2010 (PDF) and 2013
QM/EFP dispersion (PDF)
Extension of EFP to macromolecules
Description of electrostatic interactions in EFP
EFP for strong H-bonds: parameters transferability
EFP library project
Collection of EFP potentials
Libefp paper, parallelization of Libefp
QM/EFP schemes
-
EOM-CC/EFP (PDF)
-
CIS/EFP (PDF)
-
CIS(D)/EFP (PDF)
FMO projects
We also develop the fragment molecular orbital (FMO) method, in collaboration with Dmitry Fedorov and Kazuo Kitaura (AIST, Japan). FMO is a fully quantum fragmentation technique in which one performs fragment calculations in the electrostatic field of other fragments, mutually self-consistent with each other.